
Blog
Regulatory Data and Info Management for Med Device Success
With EU MDR changing the plain field of med devices to a whole a new level, other countries are following similar path of increasing demands in product safety, traceability, performance areas.
Companies not organized on information management with a system will end up duplicating lot of activities leading to loss of productivity and increase risk of non-compliance.
There are 3 categories of Challenges grouped by:
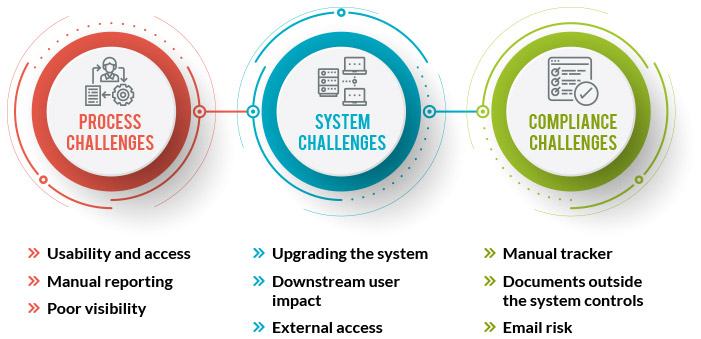
Process Challenges:
Training must be required to simplify the complexity of multiple system & user interface. As a result, users work outside of the system using local file shares or email to collaborate. To check the work status and to create report most of the companies depends on separate reporting tool or manual spreadsheets.
System Challenges:
Medical device regulation is much versatile. Upgrading a system with updated integrations is challenging and expensive as well. Systems that lie behind corporate firewalls are difficult to outsource to business partners or service providers.
Compliance Challenges:
Users develop manual tracking spreadsheets when planning & tracking capabilities aren’t part of a content management system. Many times users share information and documents via email that cause inconsistent use of document template and it is much difficult to Re-import.
Importance of a RIM system
Medical device registrations differ from country to country, with the difference in FDA’s 510K (Class I and II devices) and Premarket Approval (Class III devices) processes from the EU’s CE Marking process. The current regulatory environment in which the medical devices and diagnostics companies are competing is complex.
There is a higher demand for regulatory and compliance information required to support submissions 510 (k), PMA, De Novo and HDE.A cloud based RIM can effectively help management of product registration, commitments and regulatory submissions to medical device and diagnostics companies. This unified, single-source system provides a global user base with real-time information necessary to ensure the quality of the product and the registration.
Following capabilities should be present in an effective regulatory information management system
- Identifies device-specific global regulatory requirements.
- Assembles product information into country-specific TF template.
- Controls the dossier configurations for internal review and external review.
- Manages changes and revisions to TFs and product information.
- Generates compliant submission documents in various HA/NB formats.
- Tracks & retains submission status& manage commitments.
- Decreases effort and calendar time to replicate regulatory submission between products and regulatory agencies.
- Provides clear oversight of original and lifecycle submissions.
RIM in future:
With the versatile and novel reporting requirements set out by regulatory authorities, the plea to need Regulatory Information Management System (RIMS) is emerging brassier. The medical device market is progressively adopting regulations for UDI, eIFU, electronic submissions, others. Making these things happen without a proper RIM system is very challenging.
At the same time, Medical device companies themselves are now realizing the strategically important role that various forms of product data could play in the future; enhancing new productivity, efficiency and create competitive differentiation.
Get the latest updates from DDi
Explore Topics
Recent Blogs
 Navigating the Global eIF…In Regulations
Navigating the Global eIF…In Regulations Agentic AI in RegOps: Mov…In Automation & AI
Agentic AI in RegOps: Mov…In Automation & AI- Technical File Compilatio…In Automation & AI



CONNECT WITH US

Let's talk about how DDi can help you